Linker-Dependent Singlet Fission in Tetracene Dimers.
Korovina, N. V.; Joy, J.; Feng, X.; Feltenberger, C.; Krylov, A. I.; Bradforth, S. E.; and Thompson, M. E.
Journal of the American Chemical Society, 140(32): 10179–10190. 2018.

![Linker-Dependent Singlet Fission in Tetracene Dimers [link]](//bibbase.org/img/filetypes/link.svg "Paper") Paper
doi
link
bibtex
abstract
Paper
doi
link
bibtex
abstract
@article{Korovina_2018,
doi = {10.1021/jacs.8b04401},
url = {https://doi.org/10.1021%2Fjacs.8b04401},
year = 2018,
publisher = {American Chemical Society ({ACS})},
volume = {140},
number = {32},
pages = {10179--10190},
author = {Nadezhda V. Korovina and Jimmy Joy and Xintian Feng and Cassidy Feltenberger and Anna I. Krylov and Stephen E. Bradforth and Mark E. Thompson},
title = {Linker-Dependent Singlet Fission in Tetracene Dimers},
journal = {Journal of the American Chemical Society},
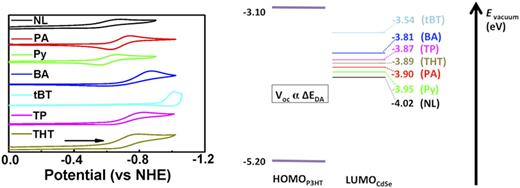
abstract = {Separation of triplet excitons produced by singlet fission is crucial for efficient application of singlet fission materials. While earlier works explored the first step of singlet fission, the formation of the correlated triplet pair state, the focus of recent studies has been on understanding the second step of singlet fission, the formation of independent triplets from the correlated pair state. We present the synthesis and excited-state dynamics of meta- and para-bis(ethynyltetracenyl)benzene dimers that are analogues to the ortho-bis(ethynyltetracenyl)benzene dimer reported by our groups previously. A comparison of the excited-state properties of these dimers allows us to investigate the effects of electronic conjugation and coupling on singlet fission between the ethynyltetracene units within a dimer. In the para isomer, in which the two chromophores are conjugated, the singlet exciton yields the correlated triplet pair state, from which the triplet excitons can decouple via molecular rotations. In contrast, the meta isomer in which the two chromophores are cross-coupled predominantly relaxes via radiative decay. We also report the synthesis and excited-state dynamics of two para dimers with different bridging units joining the ethynyltetracenes. The rate of singlet fission is found to be faster in the dimer with the bridging unit that has orbitals closer in energy to that of the ethynyltetracene chromophores.},
bibbase_note = {<img src="https://pubs.acs.org/cms/10.1021/jacs.8b04401/asset/images/medium/ja-2018-04401q_0014.gif">}
}
Separation of triplet excitons produced by singlet fission is crucial for efficient application of singlet fission materials. While earlier works explored the first step of singlet fission, the formation of the correlated triplet pair state, the focus of recent studies has been on understanding the second step of singlet fission, the formation of independent triplets from the correlated pair state. We present the synthesis and excited-state dynamics of meta- and para-bis(ethynyltetracenyl)benzene dimers that are analogues to the ortho-bis(ethynyltetracenyl)benzene dimer reported by our groups previously. A comparison of the excited-state properties of these dimers allows us to investigate the effects of electronic conjugation and coupling on singlet fission between the ethynyltetracene units within a dimer. In the para isomer, in which the two chromophores are conjugated, the singlet exciton yields the correlated triplet pair state, from which the triplet excitons can decouple via molecular rotations. In contrast, the meta isomer in which the two chromophores are cross-coupled predominantly relaxes via radiative decay. We also report the synthesis and excited-state dynamics of two para dimers with different bridging units joining the ethynyltetracenes. The rate of singlet fission is found to be faster in the dimer with the bridging unit that has orbitals closer in energy to that of the ethynyltetracene chromophores.
Manipulating Triplet Yield through Control of Symmetry-Breaking Charge Transfer.
Das, S.; Thornbury, W. G.; Bartynski, A. N.; Thompson, M. E.; and Bradforth, S. E.
The Journal of Physical Chemistry Letters, 9(12): 3264–3270. 2018.
 Paper
doi
link
bibtex
abstract
Paper
doi
link
bibtex
abstract
@article{Das_2018,
doi = {10.1021/acs.jpclett.8b01237},
url = {https://doi.org/10.1021%2Facs.jpclett.8b01237},
year = 2018,
publisher = {American Chemical Society ({ACS})},
volume = {9},
number = {12},
pages = {3264--3270},
author = {Saptaparna Das and William G. Thornbury and Andrew N. Bartynski and Mark E. Thompson and Stephen E. Bradforth},
title = {Manipulating Triplet Yield through Control of Symmetry-Breaking Charge Transfer},
journal = {The Journal of Physical Chemistry Letters},
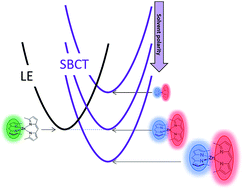
abstract = {The efficiency of an organic solar cell depends on the efficacy of exciton diffusion and dissociation processes, and this can be enhanced by reducing the exciton binding energy and increasing the exciton lifetime. Zinc chlorodipyrrin (ZCl) complexes exhibit reduced exciton binding energy due to ultrafast generation of intramolecular charge transfer (ICT) states via symmetry-breaking charge transfer in polar media. This Letter explores the fate of the ICT states using nanosecond transient absorption. In cyclohexane, ZCl undergoes intersystem crossing to produce triplets with ∼8 ns time constant (∼30% yield), and no ICT states are generated. However, in more polar solvents, triplets are generated within 1 ns via ICT state recombination with ∼3 times higher yield than produced via ISC. This high triplet yield in toluene (89%) and acetonitrile (76%) via ICT state recombination is a beneficial pathway to spin-protect the excited-state decay for additional charge generation from triplet excited states.},
bibbase_note = {<img src="https://pubs.acs.org/cms/10.1021/acs.jpclett.8b01237/asset/images/medium/jz-2018-01237h_0007.gif">}
}
The efficiency of an organic solar cell depends on the efficacy of exciton diffusion and dissociation processes, and this can be enhanced by reducing the exciton binding energy and increasing the exciton lifetime. Zinc chlorodipyrrin (ZCl) complexes exhibit reduced exciton binding energy due to ultrafast generation of intramolecular charge transfer (ICT) states via symmetry-breaking charge transfer in polar media. This Letter explores the fate of the ICT states using nanosecond transient absorption. In cyclohexane, ZCl undergoes intersystem crossing to produce triplets with ∼8 ns time constant (∼30% yield), and no ICT states are generated. However, in more polar solvents, triplets are generated within 1 ns via ICT state recombination with ∼3 times higher yield than produced via ISC. This high triplet yield in toluene (89%) and acetonitrile (76%) via ICT state recombination is a beneficial pathway to spin-protect the excited-state decay for additional charge generation from triplet excited states.
Exploring Redox Properties of Aromatic Amino Acids in Water: Contrasting Single Photon vs Resonant Multiphoton Ionization in Aqueous Solutions.
Roy, A.; Seidel, R.; Kumar, G.; and Bradforth, S. E.
The Journal of Physical Chemistry B, 122(14): 3723–3733. 2018.
 Paper
doi
link
bibtex
abstract
Paper
doi
link
bibtex
abstract
@article{Roy_2018,
doi = {10.1021/acs.jpcb.7b11762},
url = {https://doi.org/10.1021%2Facs.jpcb.7b11762},
year = 2018,
publisher = {American Chemical Society ({ACS})},
volume = {122},
number = {14},
pages = {3723--3733},
author = {Anirban Roy and Robert Seidel and Gaurav Kumar and Stephen E. Bradforth},
title = {Exploring Redox Properties of Aromatic Amino Acids in Water: Contrasting Single Photon vs Resonant Multiphoton Ionization in Aqueous Solutions},
journal = {The Journal of Physical Chemistry B},
abstract = {Direct measurements of the valence ionization energies and the reorganization energies of the three aromatic amino acids, l-tyrosine, l-tryptophan, and l-phenylalanine, in aqueous solution using the liquid microjet technique and two different photoemission methods—X-ray photoelectron spectroscopy (XPS) at 175 eV photon energy and resonant two-photon ionization (R2PI) using 2 × 267 nm (2 × 4.64 eV) UV laser light—are reported. l-Tryptophan has the lowest vertical ionization energy, 7.3 eV, followed by tyrosine (7.8 eV) and phenylalanine (∼8.7 eV). Essentially, no variation in recovered orbital energies is observed comparing near threshold ionization to X-ray ionization. Superior sensitivity of the (background-free) R2PI scheme for solutions with very low solute concentration (<2 mM) is demonstrated in contrast to the single-photon XPS measurements, which often requires solute concentrations of 0.1–1 molar. This higher sensitivity along with chemical selectivity of the R2PI technique can be exploited for both spectroscopic assignment and as an analytical tool. The nature of the adiabatic ionization energy for the three aromatic amino acids has been explored by the R2PI approach and by empirically formulating the correlation between the estimated ionization onset with electronic and nuclear relaxation on the excited state surface. Our results have implications for understanding one-electron transfer within enzymes and in redox situations where (ir)reversible deprotonation occurs such as those manifest in the biochemistry of oxidation damage.},
bibbase_note = {<img src="https://pubs.acs.org/cms/10.1021/acs.jpcb.7b11762/asset/images/medium/jp-2017-11762c_0007.gif">}
}
Direct measurements of the valence ionization energies and the reorganization energies of the three aromatic amino acids, l-tyrosine, l-tryptophan, and l-phenylalanine, in aqueous solution using the liquid microjet technique and two different photoemission methods—X-ray photoelectron spectroscopy (XPS) at 175 eV photon energy and resonant two-photon ionization (R2PI) using 2 × 267 nm (2 × 4.64 eV) UV laser light—are reported. l-Tryptophan has the lowest vertical ionization energy, 7.3 eV, followed by tyrosine (7.8 eV) and phenylalanine (∼8.7 eV). Essentially, no variation in recovered orbital energies is observed comparing near threshold ionization to X-ray ionization. Superior sensitivity of the (background-free) R2PI scheme for solutions with very low solute concentration (<2 mM) is demonstrated in contrast to the single-photon XPS measurements, which often requires solute concentrations of 0.1–1 molar. This higher sensitivity along with chemical selectivity of the R2PI technique can be exploited for both spectroscopic assignment and as an analytical tool. The nature of the adiabatic ionization energy for the three aromatic amino acids has been explored by the R2PI approach and by empirically formulating the correlation between the estimated ionization onset with electronic and nuclear relaxation on the excited state surface. Our results have implications for understanding one-electron transfer within enzymes and in redox situations where (ir)reversible deprotonation occurs such as those manifest in the biochemistry of oxidation damage.
Symmetry-Breaking Charge Transfer in Boron Dipyridylmethene (DIPYR) Dimers.
Golden, J. H.; Estergreen, L.; Porter, T.; Tadle, A. C.; R., D. S. M.; Facendola, J. W.; Kubiak, C. P.; Bradforth, S. E.; and Thompson, M. E.
ACS Applied Energy Materials, 1(3): 1083–1095. 2018.
 Paper
doi
link
bibtex
abstract
Paper
doi
link
bibtex
abstract
@article{Golden_2018,
doi = {10.1021/acsaem.7b00214},
url = {https://doi.org/10.1021%2Facsaem.7b00214},
year = 2018,
publisher = {American Chemical Society ({ACS})},
volume = {1},
number = {3},
pages = {1083--1095},
author = {Jessica H. Golden and Laura Estergreen and Tyler Porter and Abegail C. Tadle and Daniel Sylvinson M. R. and John W. Facendola and Clifford P. Kubiak and Stephen E. Bradforth and Mark E. Thompson},
title = {Symmetry-Breaking Charge Transfer in Boron Dipyridylmethene ({DIPYR}) Dimers},
journal = {{ACS} Applied Energy Materials},
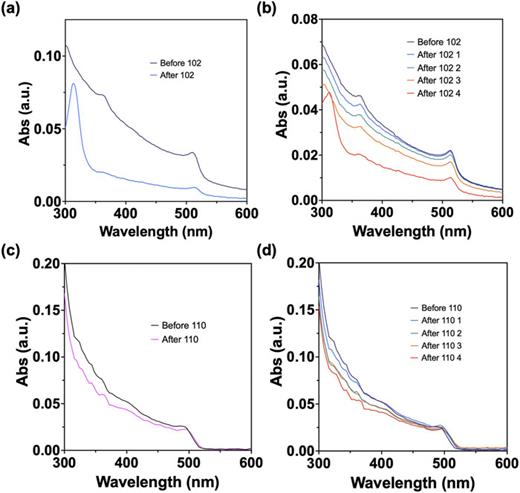
abstract = {We recently reported the photophysical properties of boron dipyridylmethene (DIPYR) dyes, a class of intensely fluorescent pyridine-based chromophores, which are structural analogues of both acenes and BODIPYs. In this work, we endeavored to explore the properties of DIPYR dimers. The synthesis and characterization of two novel homoleptic meso-linked dimers of boron dipyridylmethene dyes, bis-DIPYR and bis-α-DIPYR, are herein reported. Their structural, electrochemical, and photophysical properties have been probed using both steady-state and time-resolved techniques including femtosecond and nanosecond transient absorption spectroscopies. Of particular focus are the excited-state photophysical dynamics of the dimers, which are studied in several solvents of varying polarity, from methylcyclohexane to acetonitrile. It was found that both dimers undergo symmetry-breaking charge transfer within 3 ps of photoexcitation, forming a radical anion and radical cation, which were observed using transient absorption and confirmed by spectroelectrochemical characterization. Further, it was found that the emitting species is the symmetry-broken state, which is stable for several nanoseconds before radiative recombination to the ground state occurs. The efficiency and rapidity of symmetry breaking, even in nonpolar media, is highly promising for application of these materials to optoelectronic technologies requiring charge transfer from an excitonic state.},
bibbase_note = {<img src="https://pubs.acs.org/cms/10.1021/acsaem.7b00214/asset/images/medium/ae-2017-00214m_0012.gif">}
}
We recently reported the photophysical properties of boron dipyridylmethene (DIPYR) dyes, a class of intensely fluorescent pyridine-based chromophores, which are structural analogues of both acenes and BODIPYs. In this work, we endeavored to explore the properties of DIPYR dimers. The synthesis and characterization of two novel homoleptic meso-linked dimers of boron dipyridylmethene dyes, bis-DIPYR and bis-α-DIPYR, are herein reported. Their structural, electrochemical, and photophysical properties have been probed using both steady-state and time-resolved techniques including femtosecond and nanosecond transient absorption spectroscopies. Of particular focus are the excited-state photophysical dynamics of the dimers, which are studied in several solvents of varying polarity, from methylcyclohexane to acetonitrile. It was found that both dimers undergo symmetry-breaking charge transfer within 3 ps of photoexcitation, forming a radical anion and radical cation, which were observed using transient absorption and confirmed by spectroelectrochemical characterization. Further, it was found that the emitting species is the symmetry-broken state, which is stable for several nanoseconds before radiative recombination to the ground state occurs. The efficiency and rapidity of symmetry breaking, even in nonpolar media, is highly promising for application of these materials to optoelectronic technologies requiring charge transfer from an excitonic state.
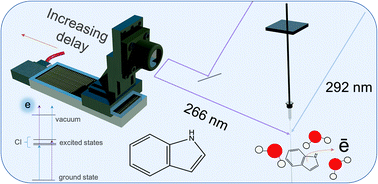
The influence of aqueous solvent on the electronic structure and non-adiabatic dynamics of indole explored by liquid-jet photoelectron spectroscopy.
Kumar, G.; Roy, A.; McMullen, R. S.; Kutagulla, S.; and Bradforth, S. E.
Faraday Discussions, 212: 359–381. 2018.
 Paper
doi
link
bibtex
abstract
4 downloads
Paper
doi
link
bibtex
abstract
4 downloads
@article{Kumar_2018,
doi = {10.1039/c8fd00123e},
url = {https://doi.org/10.1039%2Fc8fd00123e},
year = 2018,
publisher = {Royal Society of Chemistry ({RSC})},
volume = {212},
pages = {359--381},
author = {Gaurav Kumar and Anirban Roy and Ryan S. McMullen and Shanmukh Kutagulla and Stephen E. Bradforth},
title = {The influence of aqueous solvent on the electronic structure and non-adiabatic dynamics of indole explored by liquid-jet photoelectron spectroscopy},
journal = {Faraday Discussions},
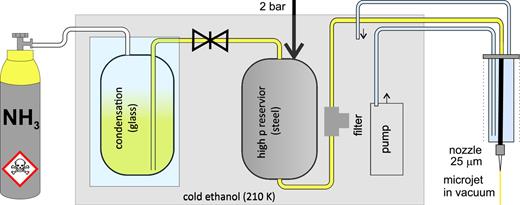
abstract = {Understanding how the electronic structure of an aqueous solute is intricately bound up with the arrangement of a host liquid provides insight into how non-adiabatic photochemistry takes place in the condensed phase. For example, the presence of water provides additional solute–solvent interactions compared to non-polar solvents: changing the stability of ionized products and modifying the energies of low-lying excited valence states, as well as moving the point of intersection between potential surfaces. Thus, the locations and topography of conical intersections between these surfaces also change. The overall impact of the aqueous environment can be to modify the intricate photochemical and non-radiative pathways taking place after photoexcitation. Time-resolved photoelectron spectroscopy (TRPES) in a liquid micro-jet is implemented here to investigate the influence of water on the electronic structure and dynamics of indole, the chromophore of the amino acid tryptophan. TRPES is used to establish ultrafast relaxation pathways that vary as a function of excitation wavelength. In our experiment, aqueous indole was excited with femtosecond pulses centered at 292 nm and 266 nm. The vertical excitation energy of aqueous indole is extracted and found to be lowered by 0.5 eV in water relative to the gas phase. In the TRPES study, the spectral signature of 1La and evidence of solvated electron formation on an ultrafast timescale are observed. Our data also points to a possible contribution of the dissociative πσ* state, which can be accessed by a conical intersection (CI) with the 1La state.},
bibbase_note = {<img src="https://pubs.rsc.org/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C8FD00123E&imageInfo.ImageIdentifier.Year=2018">}
}
Understanding how the electronic structure of an aqueous solute is intricately bound up with the arrangement of a host liquid provides insight into how non-adiabatic photochemistry takes place in the condensed phase. For example, the presence of water provides additional solute–solvent interactions compared to non-polar solvents: changing the stability of ionized products and modifying the energies of low-lying excited valence states, as well as moving the point of intersection between potential surfaces. Thus, the locations and topography of conical intersections between these surfaces also change. The overall impact of the aqueous environment can be to modify the intricate photochemical and non-radiative pathways taking place after photoexcitation. Time-resolved photoelectron spectroscopy (TRPES) in a liquid micro-jet is implemented here to investigate the influence of water on the electronic structure and dynamics of indole, the chromophore of the amino acid tryptophan. TRPES is used to establish ultrafast relaxation pathways that vary as a function of excitation wavelength. In our experiment, aqueous indole was excited with femtosecond pulses centered at 292 nm and 266 nm. The vertical excitation energy of aqueous indole is extracted and found to be lowered by 0.5 eV in water relative to the gas phase. In the TRPES study, the spectral signature of 1La and evidence of solvated electron formation on an ultrafast timescale are observed. Our data also points to a possible contribution of the dissociative πσ* state, which can be accessed by a conical intersection (CI) with the 1La state.




![How Does Mg2+(aq) Interact with ATP(aq)? Biomolecular Structure through the Lens of Liquid-Jet Photoemission Spectroscopy [link]](http://bibbase.org/img/filetypes/link.svg "Paper") Paper
doi
link
bibtex
abstract
1 download
Paper
doi
link
bibtex
abstract
1 download